

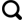
0 引 言
研究证实,气体水合物(如CH4、CO2)在处于冰点温度以下的非平衡状态时具有一种异常的稳定现象,其分解速率极其缓慢,该现象被称为水合物的“自保护”效应 [8,9,10]。了解水合物“自保护”效应的产生机制,对于利用水合物法进行天然气运输和二氧化碳运输、封存具有极其重要的指导意义。目前,多数学者认为,负温区内“冰壳”的出现与水合物“自保护”效应产生有着十分重要的关系。STERN L等先后通过甲烷水合物在193~290 K[11]、190~273 K[12]的分解实验,分析了冰对于不同类型水合物“自保护”效应的影响。TAKEYA等[13,14]先后测定了不同温度范围内甲烷水合物“自保”条件下的分解速率,提出负温条件下冰的形成是“自保护”效应产生的根本原因[13]。而后又通过研究水合物颗粒大小以及冰壳结构对水合物“自保护”效应的影响[15,16,17],进一步指出“自保护”效应的产生取决于冰壳的作用以及主—客体分子间的相互作用[15,16,17]。除此之外,FALENTY A等[18]利用共焦扫描等手段研究了二氧化碳水合物表面结构和冰壳“缺陷”在“自保护”效应形成中的作用。GREGOR等[19]研究了“自保护”初期甲烷水合物冰层的形成以及溶解的微观过程。展静等[20,21]研究了不同冰颗粒粒径对于冰点以下甲烷水合物“自保护”效应的影响。由上看出,此前大多数研究者认为,在负温条件下产生的冰壳是气体水合物“自保护”效应形成的主控因素。近期,TAKEYA K等[22]通过观察甲烷水合物“自保护”期间吸光系数以及过冷水的状态变化,提出“自保护”效应现象产生的另一种可能原因:水合物分解初期周围液态水处于过冷态,水合物所处的环境温度非常低,进而维持水合物稳定所需气体压力可达到常压,使气体水合物进入“自保护”状态。
综上看出,虽然国内外学者针对水合物“自保护”效应开展了大量宏观以及微观实验研究,但是目前对于产生这一现象的原因尚未形成完整的阐释理论。因此,本研究在不同条件下形成CO2水合物,并从水合物含气量及十二烷基硫酸钠(SDS)形成反应促进剂对“自保护”效应的影响规律2个角度出发,通过一系列实验,测定不同条件下“自保护”态水合物分解释放气体规律,从宏观实验现象角度推断微观“冰壳”、“过冷水”作用因素对二氧化碳水合物“自保护”效应产生机制的贡献程度。
1 实验装置及实验方法
1.1 实验装置
如图1所示,本实验装置由供气系统、反应系统、气体流量测试系统和数据采集系统4部分组成。供气系统由316不锈钢制储气瓶组成,其内盛有二氧化碳气体;反应系统由高压反应釜、液浴槽和温度探头(-20~30 ℃,±0.1)组成,反应釜容积为160.614 mL(含储气瓶与反应釜间的管线),釜内设置有磁力搅拌器,反应釜前后设有可视窗,通过可视窗可以观察整个实验过程中反应釜内的变化。反应釜置于空气浴之中,反应釜内温度和空气浴的温度分别通过2台超低温循环冷浴(德国Julabo)进行控制,冷浴的控温范围是 100~250 ℃,控温精度为±0.1 ℃。实验所用数据显示CO2气体流量计测量范围为0~5 L/min,分辨率为0.001 L/min,精度为±0.12%。数据采集系统可以分别采集实验过程中的温度、压力及流量数据,数据采集间隔时间为5 s,所有数据由电脑终端自动显示和储存。
1.2 实验方法
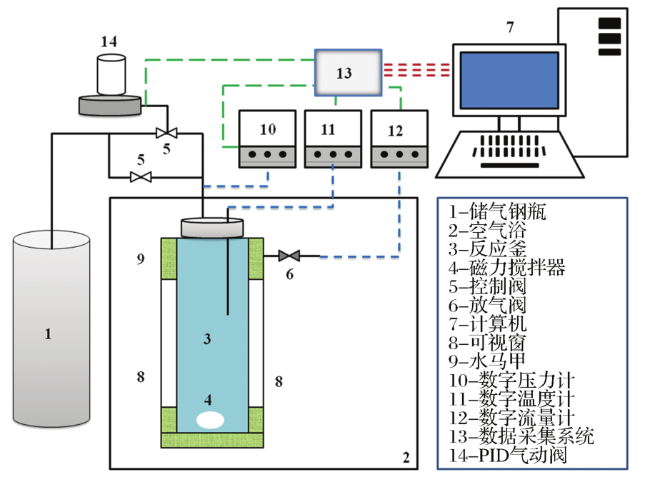
采用恒压下均匀降温法分别在不同温度下于超纯水(电阻率>18.00 MΩ·cm)和SDS(1%质量比)溶液中形成CO2水合物。总共设置4组水合物形成的目标控制温度,分别为8.5 ℃、6.5 ℃、3.5 ℃和0.5 ℃,利用CSMGem软件(由美国科罗拉多矿业学院天然气水合物中心开发),计算得到上述温度对应的平衡压力分别为3.67 MPa、2.77 MPa、1.89 MPa和1.32 MPa。
实验前,用手动注射泵将体积为104 mL的超纯水或SDS溶液注入反应釜,空气浴将实验系统温度保持在20 ℃,冷浴将反应釜初始温度维持在高于设计温度2.5 ℃。通过PID阀向釜内加压之前,先手动注压0.5 MPa,后迅速卸至大气压,重复3次,排除釜内多余空气。之后打开磁力搅拌器并设置转速为200转/min,通过PID电磁阀以间隔0.5 MPa的阶梯式加压法,将釜内压力加至设计温度对应的平衡压力值。使反应系统在此压力和温度下稳定12 h以上,使CO2气体充分溶解。然后,以3 ℃/h的速率将釜温下降4 ℃并稳定维持24 h以上,使水合物在1.5 ℃过冷度下成核并充分生长。后关闭电磁阀,将釜温在30 min内迅速降至 6 ºC并维持10 h,充分冻结已形成的水合物。最后,通过手动阀放掉釜内多余气体使水合物进入常压下的“自保护”状态,同时接通气体流量计(MF4003型,常压)并以5 s的时间间隔同步测量气体流速,并使水合物在“自保护”状态下维持5 h以上,再以3 ºC/h的速率将釜温升至实验开始时的设定值,彻底分解水合物。为了确保实验结果准确,每一条件下的实验再重复2次。
实验中所用PID阀为一种电磁阀。PID控制即比例—积分—微分控制:比例(Proportion)是依据偏差大小来动作,在调节阀系统中起稳定被调参数的作用;积分(Integral)是依据偏差是否存在来动作,起消除余差作用;微分(Differential Coefficient)是依据偏差变化速度来动作,起超前调节作用。实验所用PID电磁阀是一种利用压缩空气驱动的气动阀,阀门运行中控制信号给电磁阀通电,电磁阀则打开,此时压缩空气进入气室,推动气动阀膜,然后推动阀杆,阀杆带动阀心瞬间打开,实现由缓冲瓶向反应釜迅速补气。
2 实验结果及讨论
2.1 “自保护”效应下CO2水合物分解特征
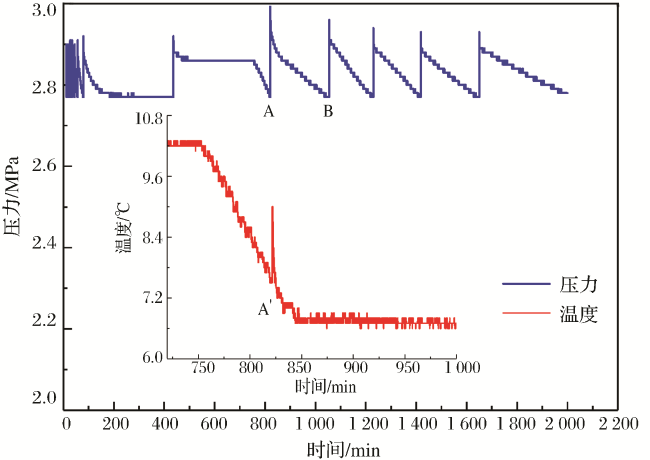
图3为6.5 ℃下生成的处于“自保护”态的水合物,在釜温逐渐上升过程中测得的水合物分解释放气体速率与时间的关系,图3(a)和图3(b)分别代表相同条件下超纯水和SDS溶液中生成水合物的分解过程,图3中横坐标零点表示水合物开始分解并释放气体时的时间点,非体系开始升温时的时间点,其他实验与此类似。由图3可看出,在釜内温度升至一定值前,大气压下被冻结的水合物不存在分解现象,即由流量计测得的气体释放速率一直为零,说明冻结态的水合物处于“自保护”状态。而当釜内温度均匀升至某一固定值后,即图中横坐标零点所对应的温度时,“自保护”态的水合物失去“自保护”能力并缓慢释放CO2气体,流量计测得的气体流速由零开始伴随时间缓慢增加。同时可以看到水合物分解一段时间以后增温速率减小,这是由于水合物分解吸热引起的。此外图3还显示,水合物释放气体过程可分3个不同的阶段,即气体释放速率迅速增大、恒定、缓慢减退的3阶段。本研究分别就每个阶段的气体释放规律进行了计算和详细分析。
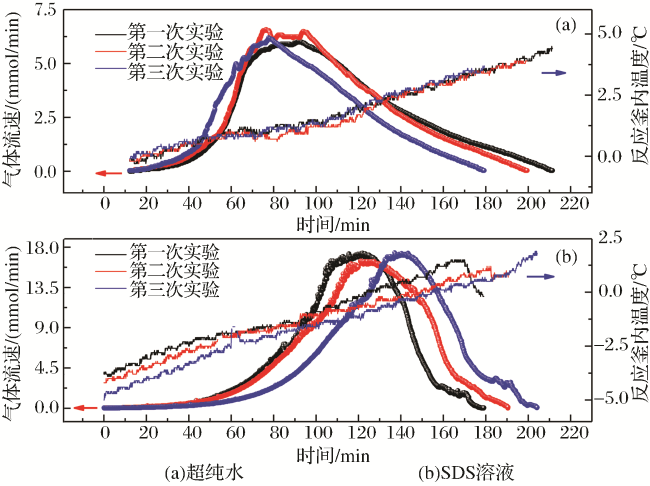
对于每个阶段,分别统计了气体流速变化与累积释放气体量间的关系。图4仅代表性地给出了在6.5 ℃下3次实验生成水合物的分解特征,其他条件下的实验与此类似。与图3类似,气体流速与累积释放气体量间的对应关系依然可分为明显的Ⅰ、Ⅱ、Ⅲ3个阶段。第Ⅰ阶段,伴随釜内温度逐渐升高水合物开始分解,气体释放速率随累积释放气体量的增加而迅速增大,直至一个最大值。第Ⅱ阶段,水合物分解释放气体的速率基本趋于稳定。第Ⅲ阶段,由于温度继续升高,剩余水合物继续分解,此时水合物分解进入最后阶段,随着累积释放气体量增加,气体释放速率逐渐减小,最终分解过程结束。由图4看出,不同阶段内气体流速变化与累积释放气体量间呈明显线性关系。因此,笔者对每个阶段内的直线斜率进行了计算。此参数表示将分解过程中每个单独阶段内测得的气体流速对累积释放气体量进行一次求导,结果对应单位为mmol/(min·mmol)。此参数实际上表征了水合物分解过程中不同阶段内气体释放速率的加速度,进而可以从物质变化规律角度表征不同阶段内的气体释放能力大小。此外,对比图3、图4可看出在同一溶液中进行的3次重复实验,其具体分解过程存在略微差异。原因是,水合物是一种非化学计量的化合物,大量实验已证实水合物成核及生长过程具有明显的随机性特征,即使在重复实验中每次实验的水分转化率也彼此不同(表1),因此重复分解过程时,每次水合物分解规律也略有差异,但呈现的总体变化规律一致。
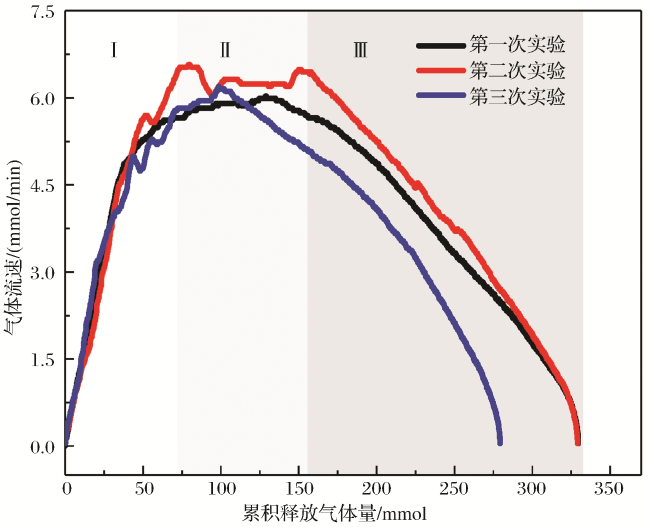
图4 6.5℃下生成的二氧化碳水合物“自保”条件下分解过程Fig.4 Decomposition process of carbon dioxide hydrate generated at 6.5 ℃ under ‘self-preservation’ condition |
表1 水合物各生成阶段耗气量Table 1 Gas consumption during each hydrate formation stage |
| 反应介质 | 实 验 次 数 | 温度/℃ | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 8.5 | 6.5 | 3.5 | 0.5 | |||||||||||
| 总耗气量 /mmol | 形成阶段 耗气量 /mmol | 冻结阶段耗气量 /mmol | 总耗气量 /mmol | 形成阶段耗气量 /mmol | 冻结阶段耗气量 /mmol | 总耗气量 /mmol | 形成阶段耗气量 /mmol | 冻结阶段耗气量 /mmol | 总耗气量 /mmol | 形成阶段耗气量 /mmol | 冻结阶段 耗气量 /mmol | |||
| 纯水 | 1 | 389.32 | 106.87 | 282.45 | 329.51 | 90.77 | 238.74 | 261.59 | 64.09 | 197.49 | 158.93 | 25.36 | 133.57 | |
| 2 | 376.88 | 97.31 | 279.58 | 329.01 | 90.44 | 238.57 | 209.97 | 28.72 | 181.25 | 158.52 | 22.48 | 136.03 | ||
| 3 | 428.88 | 122.55 | 306.32 | 279.26 | 55.56 | 223.71 | 201.48 | 27.62 | 173.87 | 157.64 | 19.97 | 137.67 | ||
| SDS溶液 | 1 | 663.30 | 147.50 | 515.80 | 1 046.09 | 535.35 | 510.74 | 1 038.06 | 607.08 | 430.98 | 275.52 | 57.03 | 218.49 | |
| 2 | 472.38 | 152.23 | 320.16 | 1 115.22 | 674.58 | 440.64 | 1 102.95 | 701.51 | 401.44 | 226.11 | 34.02 | 192.09 | ||
| 3 | 592.22 | 176.64 | 415.58 | 1 038.06 | 620.19 | 476.36 | 1 092.84 | 522.51 | 570.33 | 248.61 | 52.77 | 195.84 | ||
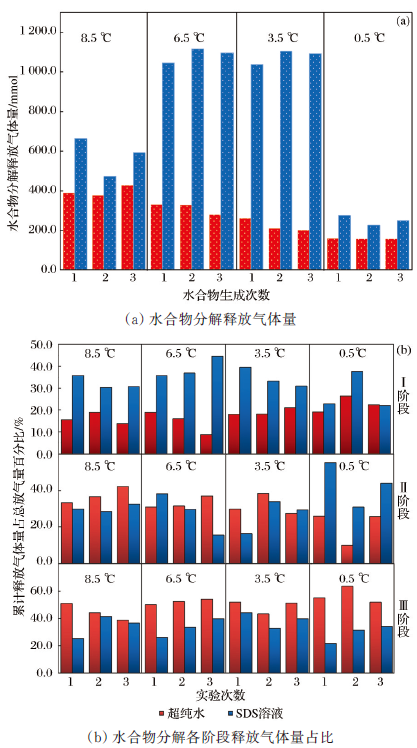
除分解过程的气体流速外,本研究还对不同条件下生成的CO2水合物的最终气体释放量进行了计算。如图5(a)所示(其中横坐标分别代表每个温度下的3次实验过程),实验温度条件会明显影响水合物分解的最终释放气体量,这是因为不同温度条件影响水合物形成过程的最终含气量。SDS溶液中,6.5 ℃、3.5 ℃下生成的水合物最终气体释放量最大,最低温度0.5 ℃下释放量最小。具体计算结果平均值为:8.5 ℃,575.97 mmol;6.5 ℃,1 085.95 mmol;3.5 ℃,1 077.95 mmol;0.5 ℃,250.08 mmol。纯水中,最终气体释放量伴随实验温度条件升高,明显逐渐增大。计算结果平均值分别为:8.5 ℃,398.36 mmol;6.5 ℃,312.59 mmol;3.5 ℃,224.34 mmol; 0.5 ℃,158.36 mmol。对比看出,除实验温度条件会对水合物含气量产生明显影响外,SDS水合物生长促进剂的加入也会明显增加水合物最终含气量:8.5 ℃、6.5 ℃、3.5 ℃、0.5 ℃下,最终含气量分别增加至145%、347%、480%、158%。其原因是,较低温度条件有利于非规整结构(具晶格缺陷或含水分子自身)的水分子笼型结构构建[24,25],因而会降低最终形成水合物的含气量。相比之下,较高温度条件会抑制非规整笼型结构的构建[24,25],因而较高温度条件下生成的水合物最终含气量会显著提高。另外,SDS是一种阴离子型表面活性剂,可明显提升水合物形成过程反应速率及水合物最终含气量[26,27,28]。因此,SDS溶液中形成的水合物均具有较高的含气量,当水合物被迅速冻结进入“自保护”状态后,仍会维持较高含气量,最终在常压条件下的升温阶段分解时,释放较多的气体。
2.2 含气量对水合物“自保护”效应的影响
表1所列为不同实验条件下水合物形成阶段以及冻结阶段的耗气量,其中冻结阶段消耗气体量是通过计算释放气体总量与形成阶段耗气量的差值得到。由计算结果可知,水合物形成阶段纯水系统内水分转化率≤13%,SDS溶液内水分转化率≤73%。因此,各实验条件下的形成反应结束后体系中仍存在大量液态水,因而在迅速冻结使水合物进入“自保护”状态的操作中,在更大过冷度驱动力下,液态水会继续转化为CO2水合物,进一步消耗气体。另外,结果还显示,即使在迅速冻结阶段,实验温度条件也会对所形成水合物的含气量产生明显影响。纯水中,伴随实验温度升高冻结阶段耗气量呈逐渐增大趋势:0.5 ℃,平均值为135.76 mmol;3.5 ℃,平均值为184.20 mmol;6.5 ℃,平均值为233.67 mmol;8.5 ℃,平均值为289.45 mmol。SDS溶液中的规律与此类似:0.5 ℃,平均值为202.14 mmol;3.5 ℃,平均值为467.58 mmol;6.5 ℃,平均值为475.91 mmol;8.5 ℃,平均值为417.18 mmol。对比发现,SDS形成反应促进剂同样会显著增大冻结阶段的水合物再次形成时的耗气量。其原因与此前2.1节中分析的温度条件、SDS对水合物含气量影响机理一致。
“自保护”态下的水合物随温度升高会失去“自保护”功能而逐渐释放气体。如图4所示,每个完整气体释放过程中,释放速率随累积释放气体量的变化规律可分为加速、稳定、减速3个明显阶段。由于每个阶段内两者间呈明显线性关系,因而笔者对每阶段对应斜率进行了计算,其单位为mmol/(min·mmol)。计算结果本质上体现的是物质的量速率变化加速度,据此我们进而可从物质变化角度,定量地比较不同阶段内水合物分解驱动力大小。图6显示了所有实验中水合物分解过程3个阶段分解加速度变化规律。第Ⅰ阶段为水合物分解加速段,因而图6中所示计算结果均为正值。此阶段超纯水中形成的水合物在失去“自保护”功能后,分解驱动力均明显比SDS溶液中水合物的大。经过快速分解阶段后,水合物进入分解速度放缓的第Ⅱ阶段。在该阶段,2种介质内形成的水合物分解加速度差异不大且都接近于零,说明即使温度持续上升(图3),此时水合物的分解驱动力也非常弱。此后,分解进入第Ⅲ减速阶段。伴随温度不断上升(图3),“自保护”态下的水合物持续释放大量气体后,气体释放能力最终减弱。该阶段内的分解加速度计算结果均为负值。在最低温度0.5 ℃下,SDS溶液中形成水合物分解速率明显比超纯水中的减小得快。其他温度条件下,两者相差不大。其原因是,温度较低时SDS的存在明显降低了CO2在溶液中的脱溶速率,气体释放能力被大大削弱。
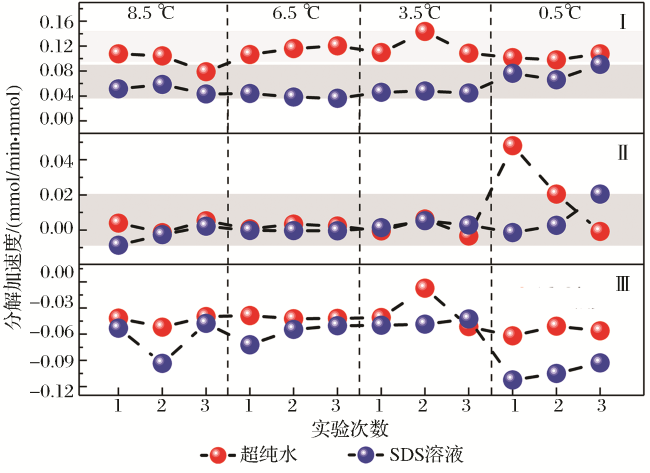
结合图5(a)和表1可看出,本研究中相同条件下SDS溶液中形成水合物的含气量均明显大于超纯水中的。所有实验中,所用介质量为固定的104 mL,因此在形成反应结束的 6 ℃冻结阶段,相对于SDS溶液,纯水系统内含有的液态水量更大(纯水、SDS溶液中水分转化率分别低于13%、73%),最终的含冰量会更高,水合物晶体周围形成的冰也会较厚。因此,如果“自保护”效应主要是由水合物外的冰引起,则纯水中形成水合物的“自保护”效果会更好。正如TAKEYA等[13]早期提出的,负温下冰的形成阻止了气体向外扩散,从而产生“自保护”效应,“自保护”效应强度应该与冰量成正比。然而,本研究的实验结果显示在第Ⅰ阶段、Ⅲ阶段与此结论相反,即第Ⅰ、Ⅲ阶段,二氧化碳水合物的“自保护”效应强度与含冰量成反比,且呈现明显重复性(结合图5,图6),这与STERN L等[11]早期的实验结果一致。另外,图6同时反映了第Ⅱ阶段2种溶液中生成水合物的“自保护”效应差别不大,即此阶段含冰量对“自保护”效应没有明显影响。同时,早期关于“自保护”效应的研究表明,并非所有的气体水合物都存在“自保护”现象,部分SII型水合物和一些大分子碳氢化合物形成的水合物不存在“自保护”现象[29],如果“自保护”效应仅仅是因为冰层引起的,显然与上述现象及本研究的实验结果不符。可见,冻结后的固体内含冰量不是水合物“自保护”效应的主控因素,尽管相关学者已经报道了“自保护”态水合物中可测到固态“冰”的存在[11,13,18]。
2.3 不同条件下生成的水合物“自保护”态下的起始分解温度
本研究中为了形成具不同含气量的水合物,除在不同温度条件下形成水合物外,还采用SDS溶液生成水合物。笔者对各升温过程中“自保护”态水合物开始分解时的温度条件进行了统计,本研究中所说“起始分解温度”,是指水合物在常压条件下经升温分解开始向外释放气体的瞬间(流量计示数从零变为非零的瞬间)所对应的反应釜内温度。实验中所有升温过程采用同一速率(3 ℃/h),进而可对不同条件下形成的“自保护”态水合物的起始分解温度进行比较。另外,升温前所有已形成的水合物均在 6 ℃冻结10 h以上,使系统中的液态水充分转化为冰(1.2实验方法)。如图7所示,水合物起始分解温度均位于0 ℃附近及以下,水合物在纯水及SDS溶液中形成,液体冰点也基本为0 ℃,因此在常压条件下的升温过程中冻结冰晶体基本不分解。
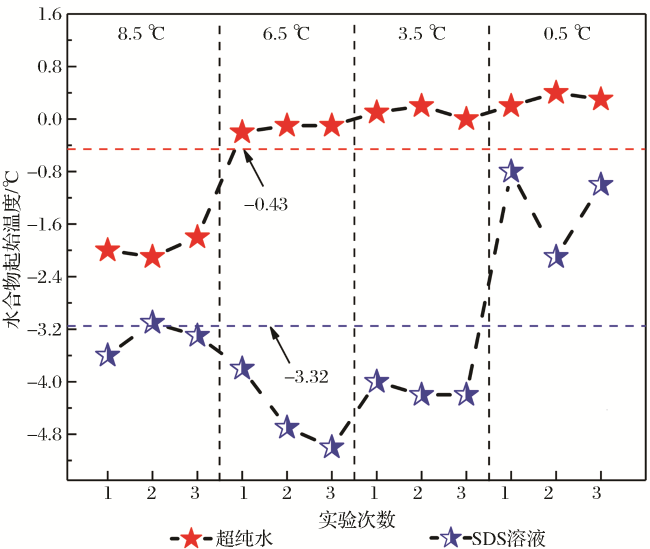
如图7所示,SDS溶液中生成水合物的起始分解温度均明显比超纯水中的低:8.5 ℃实验条件下平均值低1.37 ℃;6.5 ℃实验条件下平均值低4.37 ℃;3.5 ℃实验条件下平均值低4.23 ℃;0.5 ℃实验条件下平均值低1.6 ℃。SDS是一种动力添加剂,仅增加水合物形成速率,而不改变其生成、分解的热力学平衡条件[30,31,32]。因此,理论上SDS的加入将不会影响“自保护”态水合物分解的热平衡条件。由此可见,目前TAKEY等[22]提出的过冷水理论不能完全解释水合物“自保护”效应形成机理。他们认为,“自保护”效应的产生跟水合物初期分解时剧烈吸热生成的过冷水引起的低温条件有关。如果仅是过冷水控制“自保护”效应,那么,本研究中所有生成水合物的起始分解温度应该相同或相差不大。然而,实验结果显示,SDS溶液中生成的“自保护”态水合物起始分解温度值明显降低(图7)。
2.4 CO2水合物“自保护”现象控制因素
由以上分析看出,对于水合物“自保护”效应成因,目前存在的“冰壳”理论[11]不能在本研究中得到很好地验证,“自保护”效应更倾向由“过冷水”[22]主控,但又不是唯一因素。原因是,水合物分解时会大量吸热,因而分解初期其周围液态水处于过冷态[22]。UCHIDA等[33]通过透射电子显微镜(TEM)对CH4水合物分解溶液的冻裂模版进行测试,发现水合物分解时会在溶液中形成大量微米级、纳米级气泡(MNBs)。当溶液中的气泡为微米或纳米级时,气泡在溶液中的上升速度会明显降低[式(2)][34]。另外,伴随气泡直径减小,气泡内部压力会迅速增大[式(3)][33]。内压较高的微型气泡会在水合物分解过程中更快破裂并溶解于溶液中,使得溶液中未破裂气泡周围气体浓度显著升高[33]。气泡直径很小时,其在溶液中上升速度非常微弱[式(2)]。最终,处于“自保护”状态的水合物初步分解后晶体周围气体浓度显著增大且这种状态可长时间稳定,进而抑制水合物晶体进一步分解[35]。
V=(ρ g -ρ l )gd2/18η
P g =P l +4σ/d
式中:V为气泡在溶液中的上升速度,m/s;ρ g、ρ l分别为气泡及其周围液体的密度,kg/m3;g为重力加速度,m/s2;d为气泡直径,m;η为液体黏度,Pa·s;P g、P l分别为气泡、液体的压力,Pa;σ为水的表面张力,N/m。
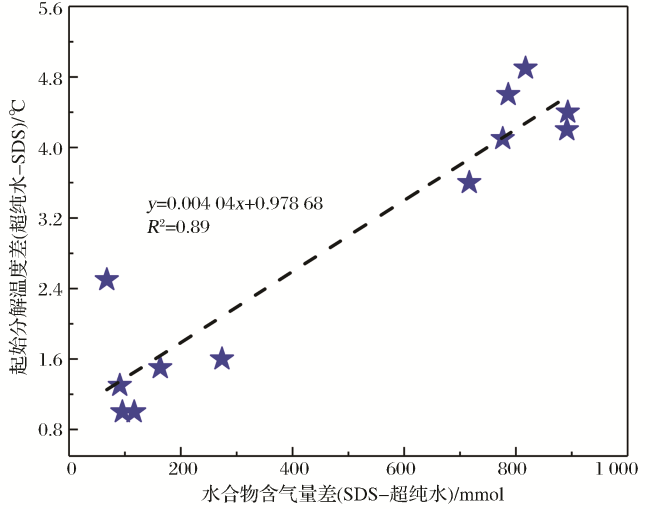
图7显示,SDS溶液中生成水合物在升温过程中的起始分解温度明显低于纯水中的起始分解温度(图7),这种差异也与冻结态水合物体系中的液态水有关。实验中对所有 6 ℃充分冻结的水合物在常压下以3 ℃/h速率升温分解,当温度达到某一值后,水合物失去“自保护”功能而开始分解,此时常压气体流量计开始测到流速,在此之前流速一直显示为零,即所有常压条件下处于冻结态水合物不存在分解现象(图3)。我们将此时对应的釜内温度定义为起始分解温度,其并非由水合物分解引起。经过卸压后,反应釜内压力达到常压,水合物—冰体系中的溶解有CO2气体的未冻液态水呈饱和态。如前文所述,与纯水相比,SDS溶液中生成水合物的含气量明显较高[图5(a),表1],因而在 6 ℃冻结过程中水合物周围最终形成的冰量较低。当缓慢升温分解“自保护”态水合物时,SDS溶液中生成的水合物—冰体系中未冻液态水量明显比纯水中的低。笔者前期的实验结果显示,温度低于3.5 ℃时,SDS不会明显影响CO2溶解度[36]。当釜温升至水合物“自保护”功能临界点之前,水合物体系中少量冰融化。相对于纯水中形成的水合物(水分转化率≤13%),SDS溶液中形成的系统(水分转化率≤73%)中由冰融化生成的未冻液态水量明显较低,进而使液态水内可溶解的CO2气体量也明显较低。因此,在升温过程中SDS溶液中形成的水合物系统中可提供溶解CO2气体分子的未冻液态水量较低,即系统对水合物晶体提供的保护能力较弱,因此SDS溶液中“自保护”态下水合物起始分解温度较低。相反,纯水中水合物含气量较低(水分转化率≤13%),冻结时生成的冰量较大,升温过程中会生成较多的未冻液态水,进而在水合物分解临界点会容纳更多的CO2气体分子,最终使得“自保护”态水合物起始分解温度比SDS溶液中的普遍较高。另外,图8显示不同实验条件下“自保护”态水合物起始分解温度差异(绝对值)与含气量差异呈明显线性关系(R 2=0.89)。进一步表明,CO2水合物“自保护”效应形成条件与其中的自由态水量直接相关。
3 结论
关于水合物“自保护”效应的产生原因,目前国际上主要存在“冰壳”、“过冷水”2种阐释机制。本文研究通过在恒压下降温的方法在不同温度下形成具有不同含气量的CO2水合物,在-6 ℃对其进行充分冻结后,采用均匀升温的方法分解“自保护”态下水合物,测量气体释放速率及水合物起始分解温度。将测得的释放速率对累积释放气体量一次求导,以此表征“自保护”态下水合物不同分解阶段内的气体释放能力,并结合测得的起始分解温度,对上述2种机制进行验证。结果发现:“自保护”态下水合物在相同升温条件下开始分解时,气体释放能力与冻结态水合物内冰量成反比;形成反应促进剂SDS(十二烷基硫酸钠)会明显降低水合物的起始分解温度。对比分析发现,“冰壳”理论无法完全解释本研究实验结果。本文提出:CO2水合物“自保护”效应更倾向于由水合物初步分解时周围存在的过冷液态水,以及液态水内溶解的微型(微米、纳米级)气泡控制。
 甘公网安备 62010202000678号
甘公网安备 62010202000678号
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}